

Understanding the core elements of Quality Management Systems
In today’s competitive marketplace, delivering high-quality products and services is essential for business success. A Quality Management System (QMS) is

5 Differences Between ISO 13485 & FDAs Medical Device QSR
Medical device manufacturers face a daunting task: navigating complex regulatory landscapes to ensure their products meet safety and quality standards.
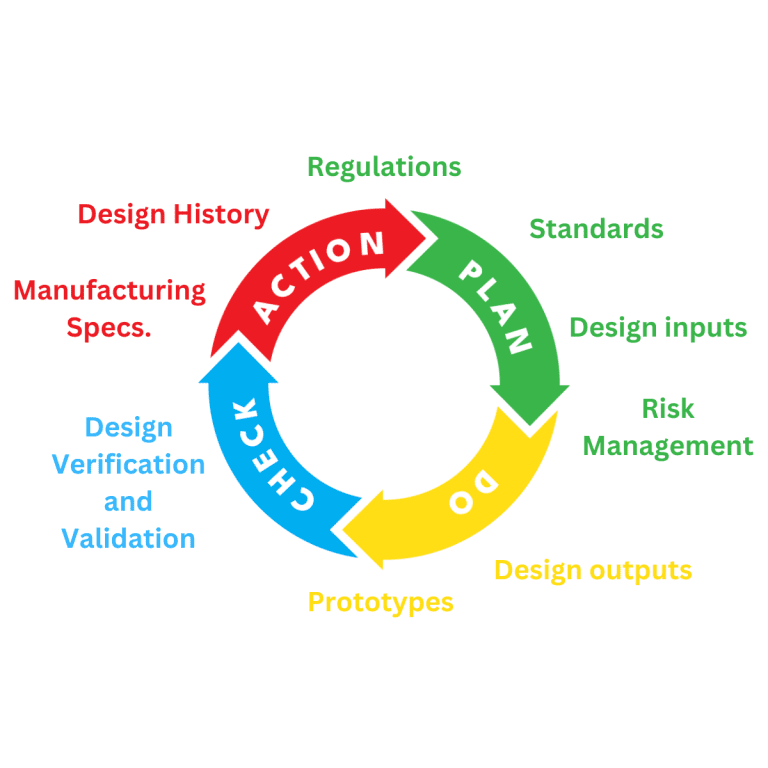
Medical Device Design and Development
Planning for the design and development of a medical device is a requirement of regulatory systems. All manufacturers of Medical
ISO 13485 – Quality Management System
The ISO 13485 standard was built around the structure of ISO 9001 which is a standard for Quality Management Systems