

Post-Market Surveillance (PMS) for Medical Devices
Learn how post-market surveillance (PMS) helps medical device manufacturers monitor safety, collect real-world evidence, maintain MDR compliance and improve devices

Medical Device Clinical Evaluation
Learn how medical device clinical evaluation demonstrates safety, clinical performance and regulatory compliance under the EU MDR and UK medical

Is My Product a Medical Device?
When developing a new and exciting product it can be hard to know if it might fall under specific regulations.
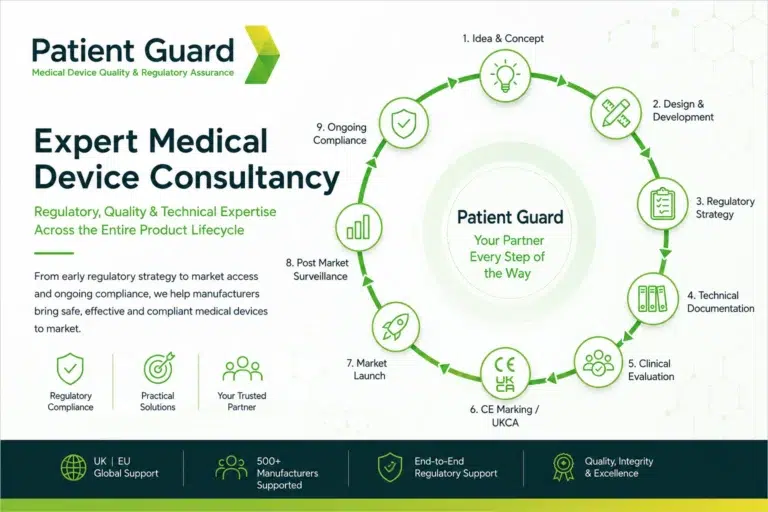
Medical Device Consultancy
Medical device consultancy helps manufacturers navigate complex regulatory, quality and technical requirements throughout the product lifecycle. From early regulatory strategy
