IEC 62366-1 Usability Engineering and its use in Medical Device Design and Development
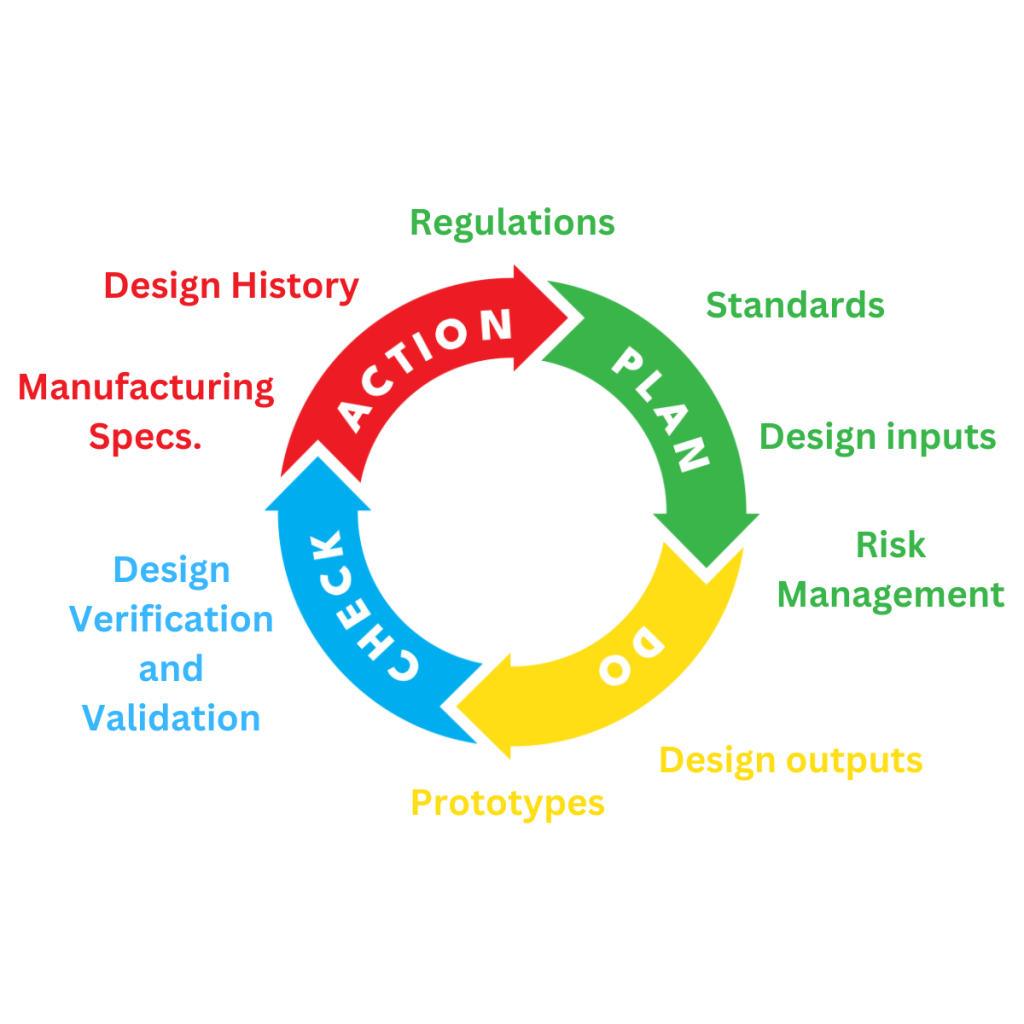
In medical device design, the efficacy, safety, and user experience is paramount. While the technical function of a device is unquestionably significant, its usability can often determine its real-world impact. This is where standards like IEC 62366-1 come into play, offering a structured approach to integrating user-centred design principles into the development process. In this blog, we’ll delve deeper into the significance of IEC 62366-1, its underlying principles, practical implementation strategies, and the broader implications for medical device innovation and patient care.
IEC 62366-1 Usability Engineering and its use in Medical Device Design and Development Read More »