MDR (EU)2017/745 Extention
In 2017 the EU introduced a new regulation for medical devices. Medical device regulation (MDR) EU 2017/745. These new regulations repeal the old EU Directives, Medical Device Directive (MDD) 93/42/EEC, Active Implantable Medical Device Directive (AIMDD) 90/385/EC. The MDR combines the requirements of medical devices and active implantable medical devices under one regulation.
Why new regulation?
The new regulations are more up to date and include provisions around newer technologies, such as software. They also introduce more stringent requirements in demonstrating compliance for manufacturers, stricter rules for Notified Bodies to follow and better regulations around traceability of medical devices.
The new regulations apply to all medical devices placed on the EU market or countries that accept CE marking, not just new medical devices.
Old Time Frames
Manufacturers were given 3 years to become compliant with the MDR. This was defined within the requirements of Article 120 in both regulations.
This was a very ambitious timeframe, unfortunately since then there have been a large number of factors that have made this timeframe unachievable. We will discuss this in further detail.
The importance of article 120 meant that devices that did not conform to the new regulations by the 2020 deadline would no longer be able to place those devices on the EU market.
Legacy Devices
With the regulations requiring all medical devices which are placed on the EU market, not just new ones, it has meant that all legacy devices must now be complaint with the new regulations in order to apply the CE mark. There are over 500,000 different types of medical devices currently placed on the EU market. This equates to hundreds of millions of medical devices and thousands of different medical device manufacturers.
For those that manufacture class Is, class Im, class IIa, class IIb and class III medical devices, they require assessment by an EU approved Notified Body, who issues CE certificates, before they can place or continue to place devices on the EU market.
In order to approve all of those medical devices within 3 years for medical devices was always going to be a challenging time frame especially given that Notified Bodies had to be assessed by the EU authorities against the requirements of MDR before they were allowed to issue CE certification against the new MDR.
Notified Bodies
As a result of the new regulations, and the new more stringent requirements on them in terms of assessment, as well as requiring specialist clinical employees relevant to the type of medical devices they were assessing, made the costs of being a Notified Body increase significantly.
Due to this, the number of Notified Bodies decreased from over 55 to less than 35. Those who remained had to go through a very strict assessment by the EU authorities in order to be granted a licence to issue CE certificates under MDR. This was a slow process, and so the number of Notified Bodies who could issue certificates under the new regulations remained low for a prolonged period of time.
It started to emerge that unless there was an increase in the number of Notified Bodies able to issue CE certificates under MDR, then there was a real risk that there maybe a shortage of medical devices in member states healthcare systems, which could have led to significant harm to the population.
COVID
By 2019/2020 the COVID pandemic took its grip of the world. Everything came to a standstill as the world shut off and we went into lockdowns. The focus turned away from regulatory compliance and a focus on medical device manufacturers manufacturers in ramping up production of face masks, ventilators and COVID tests. The regulators needed to focus on the emergency approval of COVID tests, ventilators and vaccines, and movement restrictions hampered Notified Bodies being able to carry out audits.
Due to this the EU commission decided to extend the provisions of Article 120 for another year. Meaning that medical device manufacturers now had until 2021 for achieving compliance. In hindsight this was probably too little, and a longer time frame should have been put in place.

EUDAMED
A cornerstone of the MDR regulations was the implementation of an EU wide medical device electronic system for registration, traceability and monitoring of medical devices. This system is known as EUDAMED. The system is intended to be comprised of 6 modules that will allow medical device manufacturers to register medical devices on the EUDAMED system, raise adverse incident complaints, upload vigilance and PMS data.
The system is intended to allow Notified Bodies to upload CE certificates, and monitor organisations they are involved with, and to allow authorities to make EU wide decisions on medical devices based on their traceability, conformity data, PMS data. The system was intended to allow EU wide conformity and uniformity. As the current system is based on a state level process, and communication between authorities where a medical device is placed in multiple member states is poor.
Unfortunately, the implementation of the EUDAMED system is behind schedule. Currently there are only two modules active. As EUDAMED is enshrined in detail within the medical device regulations, it makes it difficult to enforce the regulations whilst the system is not fully operational.
Article 120
Given all these factors there was and remains a significant backlog of CE mark certificates that are about to run out under the old medical device directives. This means that without an extension to article 120, there was the risk that medical devices would be limited in the EU and all the horrible scenarios both medically and economically that would have caused.
Based on this the EU commission decided that CE marks under the old directives could be extended by notified bodies and medical devices still placed on the market.
The notified body that issued the AIMDD or MDD certificate may confirm in writing (after having reviewed manufacturer’s description of the (proposed) change) that the implementation of the change does not represent a significant change in design or intended purpose under MDR Article 120(3) and that the related AIMDD or MDD certificate remains valid after the date of application of the MDR, but no longer than its expiry date or 26 May 2024, whichever comes first.
This was still not enough time considering the low number of manufacturers that had gone though the MDR and IVD process, so on 20 March 2023, Regulation (EU) 2023/607 entered into force.
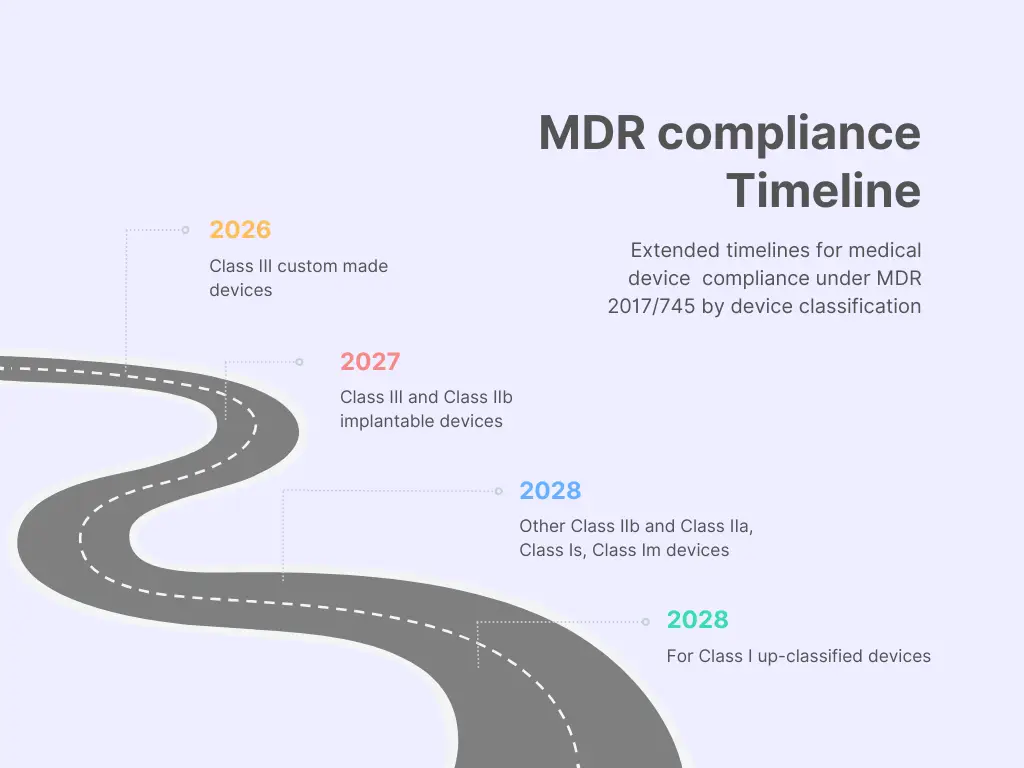
Article 120 - New Time Frames
This Regulation extends the transitional provisions of the MDR as follows:
- 2026 for class III custom made devices,
- 2027 for class III and class IIb implantable devices,
- 2028 for other class IIb, class IIa and class Is, Im devices, and
- 2028 for class I up classified devices.
The Regulation also removes the ‘sell off provision’ for both the MDR and IVDR. This means that devices already placed on the market can continue to be made available or put into service until the revised expiry of the certificate or until the shelf life of the device.
However, the regulators have stated that by 26th May 2024 medical device manufacturers must have a contract in place with a Notified Body showing that they will be moving across to MDR at some point before the end of the deadlines.
Why Manufacturers Should Stay Vigilant
While the extension offers a breathing space for notified bodies, it is imperative for medical device manufacturers not to interpret this as a relaxation of regulations. Compliance with these regulations remains non-negotiable. Manufacturers must continue to meet the stringent criteria outlined in the EU regulations to ensure that their products are safe and effective for patients. Cutting corners or delaying compliance measures not only risks regulatory repercussions but also jeopardizes patient safety and trust.
Proactive Measures: Partnering with Experts
In the face of these regulatory changes, it is crucial for medical device manufacturers to take proactive steps. Rather than viewing the extension as an opportunity to procrastinate, manufacturers should use this time wisely to fortify their compliance processes. One of the most effective ways to do this is by partnering with experienced medical device consultancies, such as Patient Guard.
Why Patient Guard?
Patient Guard is a leading medical device consultancy renowned for its expertise in regulatory compliance. By collaborating with consultants who understand the intricacies of the regulations, manufacturers can streamline their certification processes. Engaging with experts not only helps in navigating the complexities of compliance but also ensures that manufacturers stay ahead of the rush. Being proactive in seeking guidance can make a significant difference, enabling manufacturers to obtain certifications efficiently and effectively.

Upholding Excellence in Medical Device Manufacturing
The extension of Article 120 in the EU Regulation 2017/745 is a testament to the EU’s dedication to patient safety and product quality. However, for manufacturers, this extension should serve as a reminder rather than a reprieve. By proactively adhering to the regulations and seeking expert guidance from consultancies like Patient Guard, manufacturers can not only meet the compliance standards but also contribute to a healthcare landscape where patient safety and quality are paramount. Remember, compliance is not just a legal obligation; it’s a commitment to excellence, integrity, and the well-being of patients worldwide.


