ISO 14971: Understanding the International Standard for Medical Device Risk Management
ISO 14971 and the Risk Management of Medical Devices plays an integral part of demonstrating product safety throughout the life cycle of medical devices. In fact, the focus of all global medical device regulations relate to risk and how these are mitigated, to demonstrate that the clinical benefit of using the medical device outweighs the risks associated with using the medical device.

ISO 14971 Risk Management: A Step-by-Step Guide for Medical Devices
ISO 14971 is an international standard, a set of ‘state of the art’ and best practice principles adopted by medical device manufacturers. The standard sets out how risks should be identified, assessed for severity and likelihood of occurrence, applying control measures, re-assessment of the risk once control measures are in place, evaluation of residual risks introduced by control measures.
It provides a framework to follow to assess risk at each stage of the product life cycle:
- Design
- Manufacturing
- Packaging
- Transport
- Storage
- Use
- Obsolescence/disposal
The standard is a harmonised/consensus standard. This means that it should be used to demonstrate medical device compliance for the aspects of risk management in accordance with medical device regulations. This includes EU MDR 2017/745, EU IVDR 2017/746, UK MDR 2002 and FDA CFR 21.
ISO 14971 - Risk Management Plan
The first aspect to consider when implementing ISO 14971 is planning. A risk management plan should be developed to demonstrate how risk management will take place. This should be a set of instructions detailing in what form risk management activities will be conducted, the type of risk management principles that will be applied (FMEA for example). It should set out the criteria for what constitutes an acceptable risk compared to an unacceptable risk, and what the actions are based on the risk acceptability criteria.
The risk management plan should include the stages at which risk management should take place, and who should be involved in risk management activities. It should also include when and how risk management should take place in relation to post market surveillance and vigilance activities, such as if there was an adverse incident relating to the use of the medical device, that was not foreseen when originally assessing the risks of the medical device.
The aim of the plan is to ensure that medical device manufacturers reduce risks to as low as possible. Risk management is an ongoing process and documentation continually kept up to date within the risk management file.
Each medical device type should have its own risk management documentation, this is usually kept with the technical documentation in the medical device file.
ISO 14971 - Risk Identification
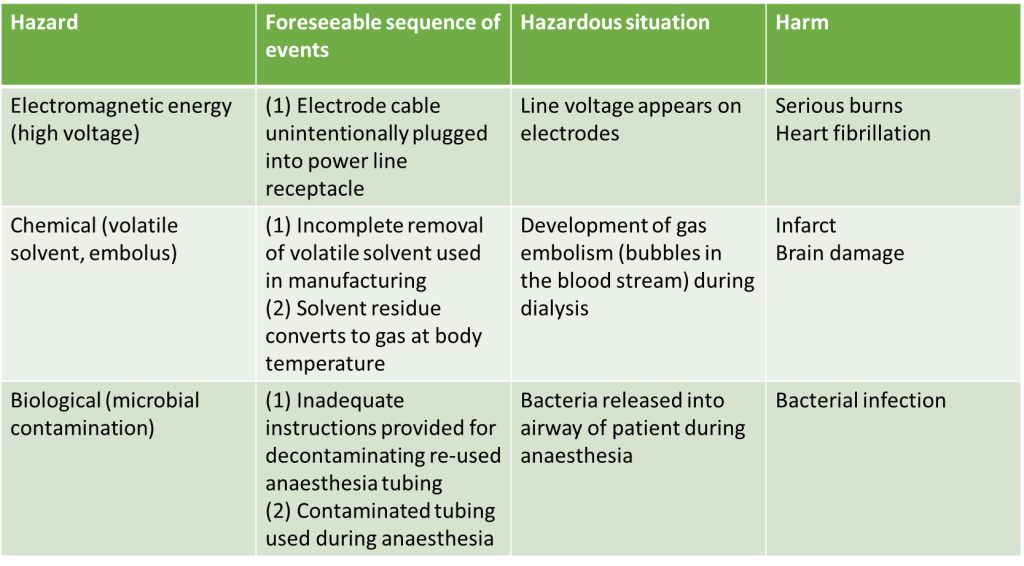
Manufacturers of medical devices are responsible for identifying all the known and foreseeable risks associated with the use of the medical device both within its intended use and misuse of the medical device, the risks should be assessed in both normal and fault conditions. Here is an example taken from Annex C of the ISO 14971 standard on how this information should be presented.
Identified Risks Example
ISO 14971 - Risk Analysis
Once risks have been identified they should be assessed based on their severity and likelihood of occurrence. They should also be assessed for acceptability based on the level of risk identified. One way this can be done is through FMEA.
Severity is given a number of 1 to 5, with 1 being the lowest risk and 5 being the highest risk and the same for likelihood of occurrence. Its important to remember that the severity of the identified risk will always be the same even after control measures have been put in place, but the likelihood of risk can be moved to a lower number once mitigated and controlled.
Risk Score Matrix
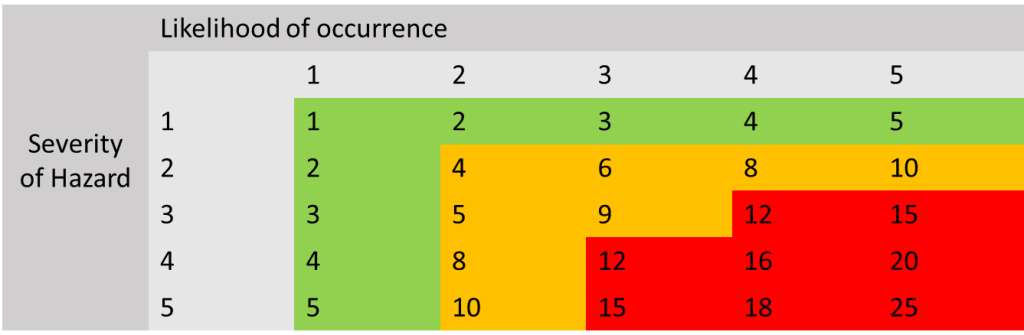
Here is an example of how risk acceptability works. The numbers in the green section are generally considered to be acceptable risks and have been reduced to as low as possible. Numbers in the orange section are medium level risks and need to be assessed to see if they can be reduced further, and if they can not then they need to be assessed to see if the risk is considered to be acceptable when weighing up the benefit of using the device. Numbers that are in the red zone need to be reduced further and are not acceptable risks, if these risks cannot be reduced further by control measures, the manufacturer needs to think about redesigning/manufacturing, changing the intended use etc. of the device to ensure the risk is removed or changed to an acceptable risk level.
Applying Risk Score to Risks Identified Example
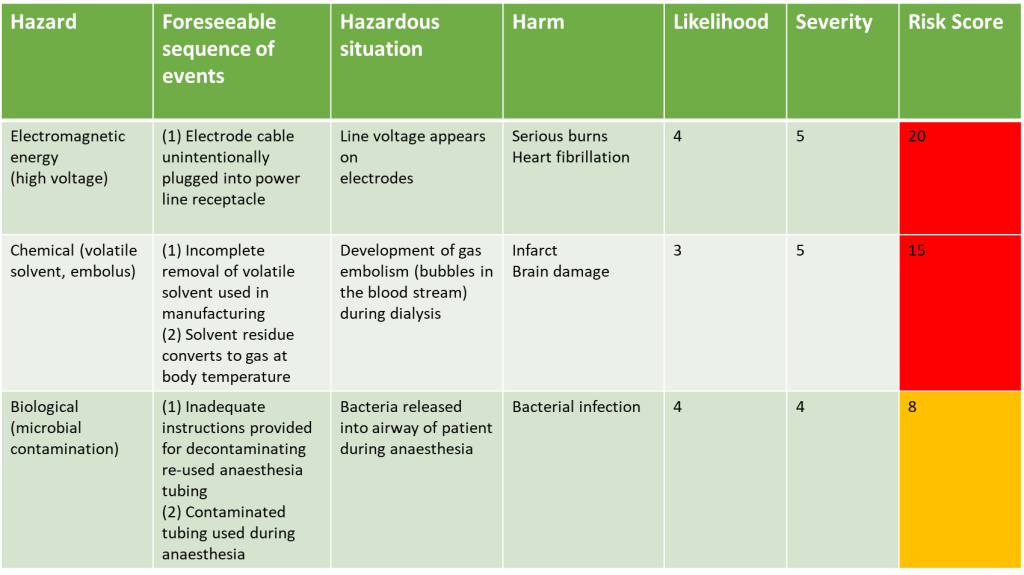
Let’s go back to our example of the identified risks, lets assume the risk management group have reviewed all the potential risks in terms of their severity and considered the likelihood of them occurring:
They would then see that the three risks identified, once risk scores have been applied, are not in the green zone, and therefore are not acceptable and need to have control measures put in place to reduce the risk to as low as possible.
ISO 14971 Risk Evaluation
Once risk control and mitigation factors have been put in place, the manufacturer should assemble the risk management team to evaluate the risk control measures to ensure that the risk has been reduced to as low as possible. They should also evaluate any residual risks that remain after the control measures have been put in place by performing the risk analysis of the control measure against the original risk identified. If the control measures do not mitigate the risk to an acceptable level, then they should take action such as redesign, changes in manufacturing and so on, and then assess the risk again.

In some cases, the implementation of a control measure may introduce new risks. In this case these new risks should be documented, and the process described above followed.
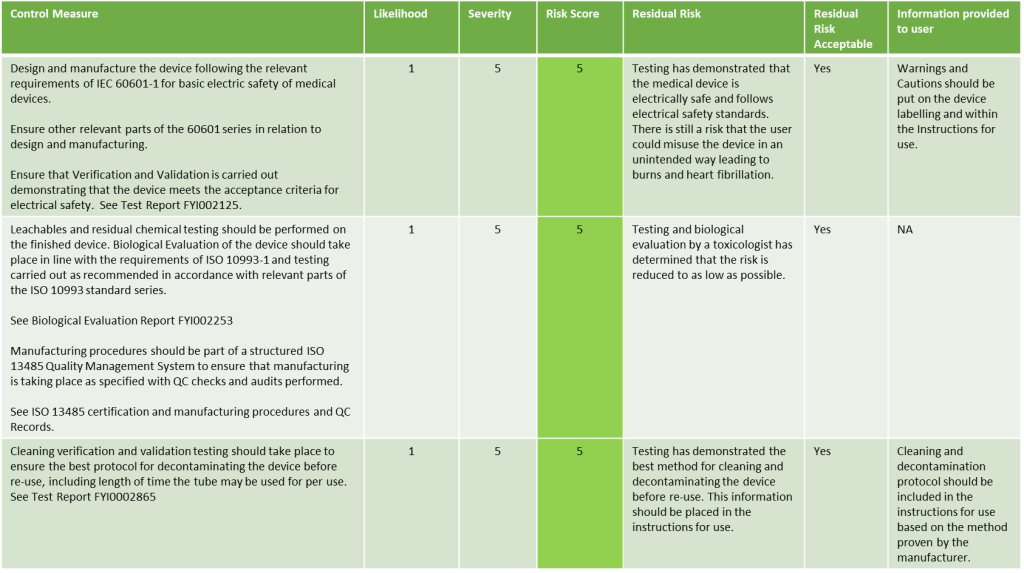
Looking at our example above, here we describe what the risk evaluation may look like:
Evaluation of Risks once Risk Control measures have been implemented Example
ISO 14971 - Risk-Benefit Assessment
Once the device is in its final finished form ready for release. The Risk Management Team should evaluate the over all risk documentation that has been generated and evaluate if the residual risks are acceptable and that all residual risk information where a risk is still possible is documented either on the medical device labelling in the form of symbols following ISO 15223-1, and in the instructions for use, providing the end user with important information on how to use the device safely.

In this review all the risks and residual risks should be assessed against the clinical intended use of the device. They should assess that the over all risks associated with the use of the device as intended, are outweighed by the benefits of using the device for its intended purpose.
ISO 14971 - Continual review
Risk Management doesn’t end when a medical device has been launched and is on the market. The risk management team should regularly check the post market surveillance data being generated from within the Quality Management System from manufacturing records, customer feedback, customer complaints, adverse incidents and review of data being generated about other similar devices placed on the market such as from competitor devices.
New unforeseen risks should be logged and assessed on an ongoing basis and control measures put in place including re-design or manufacturing changes if needed. The manufacturer should also take into account the risks associated with withdrawal or discontinuation of the medical device.
ISO 14971 - Summary
ISO 14971 is an international standard for the application of risk management to medical devices. It provides a systematic approach for identifying, evaluating, and controlling risks associated with the use of medical devices throughout their lifecycle. The standard emphasizes continuous risk assessment, ensuring that risks are reduced to acceptable levels and that residual risks are clearly communicated. It is widely used in regulatory compliance to enhance the safety and effectiveness of medical devices.
How can Patient Guard help?
We hope you found this article useful, its always important to include someone with medical device regulatory experience on your medical device risk management team, this is to help navigate any regulatory requirements or changes which may impact you and your medical devices.
Contact Patient Guard to ensure ISO 14971 compliance and robust risk management for your medical devices
Frequently Asked Questions (FAQs)
The purpose of ISO 14971 in medical devices is to provide a systematic framework for identifying, evaluating, and controlling risks throughout a device’s lifecycle, ensuring that risks are reduced to acceptable levels while demonstrating compliance with regulatory safety requirements.
ISO 14971 helps with risk management in compliance with EU MDR by providing a harmonized framework to identify, evaluate, and control risks, ensuring that medical devices meet safety and performance requirements while demonstrating that benefits outweigh risks as required by EU MDR.
The key steps in the ISO 14971 risk management process are:
- Risk Analysis: Identify hazards, estimate risks, and evaluate risk acceptability.
- Risk Control: Implement measures to reduce risks and reassess residual risks.
- Evaluation of Overall Residual Risk: Ensure all remaining risks are acceptable and benefits outweigh risks.
- Post-Market Surveillance: Continuously monitor and update the risk management file throughout the device lifecycle.

