
EUDAMED
The EU Commission has recently announced that the transition period for completing and implementing EUDAMED has been extended again. The previous target for full functionality was the second quarter of 2024. The new target date for this is now the second quarter of 2027.
EUDAMED will consist of 6 interconnected modules with a public website:
- Actors Registration
- UDI/Devices Registration
- Notified Bodies and Certificates
- Clinical Investigations and Performance Studies
- Vigilance and Post-Market Surveillance
- Market Surveillance
- EUDAMED Public (website)
EUDAMED is not yet mandatory nor required at this stage but some modules are already available and can be used voluntarily. The three modules currently in operation are; Actor Registration, UDI/Device Registration and Notified Bodies and Certificates. The remaining modules are still currently under development and will be released when EUDAMED is declared fully operational.
The proposed timelines for mandatory use of all modules are as follows:
EUDAMED (time line)
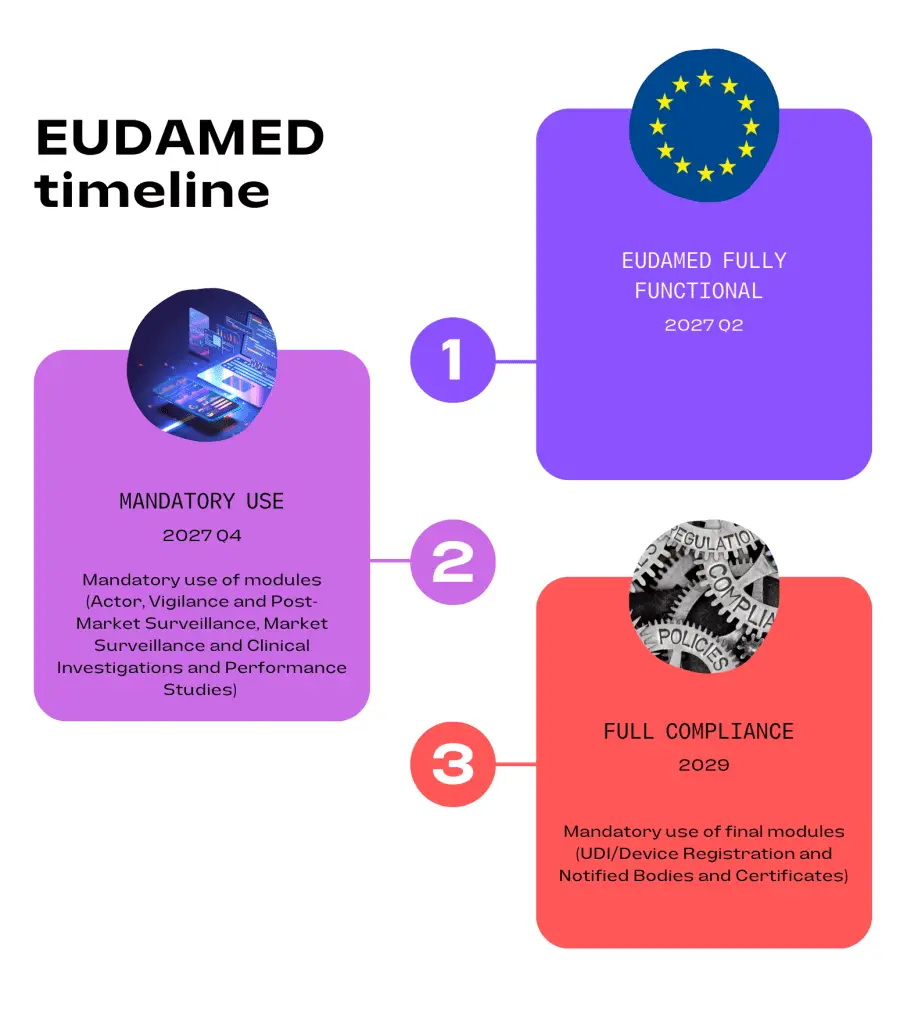
Once all modules are complete an independent audit of these will be planned and should take place in the fourth quarter of 2026. This will be followed by an official EU report then a six month transition period.
Why the delay?
Full functionality of EUDAMED was originally planned for May 2020. Extensions have been included to allow more development time for the modules and a staggered release introduced. The second release date was planned for the end of 2023 but again, postponement was required due to some modules not being developed, specifically the Clinical Investigations and Performance Studies module. The new timelines shown above may not be met due to the complexity of the modules and further delays are possible.
There are currently 40 Notified Bodies designated for regulation EU MDR 2017/745 in the EU. These NBs are responsible for auditing and issuing certificates for medical devices outside of Class I. Due to this relatively small number of approved Notified Bodies there are considerable delays in the re-certification of medical devices.
So what does this mean?
The extension to the regulations and EUDAMED database full release is not necessarily a negative impact for medical device manufacturers. This delay allows more time for adaption to the EU MDR 2017/745 and registration with the EUDAMED database. However, Patient Guard strongly recommend that manufacturers of medical devices register on EUDAMED and start to use any modules currently functional.
Current status of the European Commission on EUDAMED and functional modules
The following information is interpreted from the European Commission, Public Health website.
The three modules currently live are Actor Registration, UDI/Device Registration and Notified Bodies and Certificates.
The EU states that:
“The Commission is not in a position to require the use of Actor Registration module until EUDAMED is fully functional according to the Medial Device Regulation.”
“The European Commission is not in a position to require the use of the UDI/Devices registration module until EUDAMED is fully functional according to the Medical Device Regulation”.
“The European Commission is not in a position to require the use of the NBs & Certificates module until EUDAMED is fully functional according to the Medical Device Regulation”.
However;
Actor Registration: Every economic operator (EU and non-EU manufacturers, authorised representatives, system/procedure pack producers and importers) has to register as an actor and provide the required information.
The required information is as follows:
Declaration on information security responsibilities
All actors must upload a signed declaration on information security responsibilities.
Mandate summary document
To register, the non-EU manufacturers must have an active authorised representative and submit with the registration a mandate summary document.
UDI/Devices registration: Manufacturers can enter UDI/Device information in the EUDAMED system voluntarily.
Notified Bodies and Certificates module: NBs can register certificates and Summaries of Safety and Clinical Performance (SSCP) only on voluntary basis if all of the parties referenced in the certificates are first registered, also on a voluntary basis, in this database.
How can Patient Guard help?
Keep up to date with the latest regulatory news and updates by singing up to our news letter below, or contact us to see how using our medical device regulatory and quality assurance services can make your compliance journey easier.
FAQs
EUDAMED (European Database on Medical Devices) is a centralized database established under the EU Medical Device Regulation (MDR 2017/745) and In Vitro Diagnostic Regulation (IVDR 2017/746). It aims to enhance transparency, traceability, and oversight of medical devices within the European Union.
Why it matters: EUDAMED enables regulatory authorities, manufacturers, and the public to access vital information about medical devices, ensuring safety and compliance.
EUDAMED consists of six interconnected modules, each serving a specific purpose:
- Actor Registration: Records details of economic operators (e.g., manufacturers, authorized representatives, importers).
- UDI/Device Registration: Tracks Unique Device Identifiers (UDI) and device details.
- Notified Bodies and Certificates: Maintains information about notified bodies and issued certificates.
- Clinical Investigations and Performance Studies: Records ongoing and completed studies.
- Vigilance and Post-Market Surveillance: Monitors incidents, trends, and safety measures.
- Market Surveillance: Facilitates communication between national authorities for oversight.
Pro tip: EUDAMED integrates all regulatory processes into one platform, streamlining compliance and monitoring.
The following economic operators must register in EUDAMED:
- Manufacturers: Including those based outside the EU selling into the EU market.
- Authorized Representatives (AR): Acting on behalf of non-EU manufacturers.
- Importers: Responsible for placing devices on the EU market.
- Notified Bodies: Overseeing conformity assessments and certifications.
Key insight: Registration in EUDAMED is a critical step for compliance with the MDR and IVDR.
The UDI is a unique code assigned to medical devices, enabling easy identification and traceability. In EUDAMED, the UDI:
- Links devices to their technical documentation.
- Ensures traceability across the supply chain.
- Facilitates market surveillance and recalls if necessary.
Key takeaway: UDI registration is mandatory for all medical devices under the MDR/IVDR.
As of now, EUDAMED is being implemented in phases:
- Some modules, like Actor Registration, are live and operational.
- Other modules, including UDI/Device Registration and Vigilance, are expected to be rolled out in the coming years.
Pro tip: Stay updated on EUDAMED’s timeline to ensure timely compliance with new requirements.
Yes! Patient Guard provides expert support for EUDAMED-related activities, including:
- Registering economic operators and devices in EUDAMED.
- Assisting with UDI compliance and device data submissions.
- Acting as your EU Authorized Representative for non-EU manufacturers.
- Providing guidance on clinical investigations and vigilance reporting requirements.
Why choose Patient Guard: With extensive experience in EU MDR and IVDR compliance, we simplify EUDAMED registration and ensure your organization meets all regulatory requirements.

