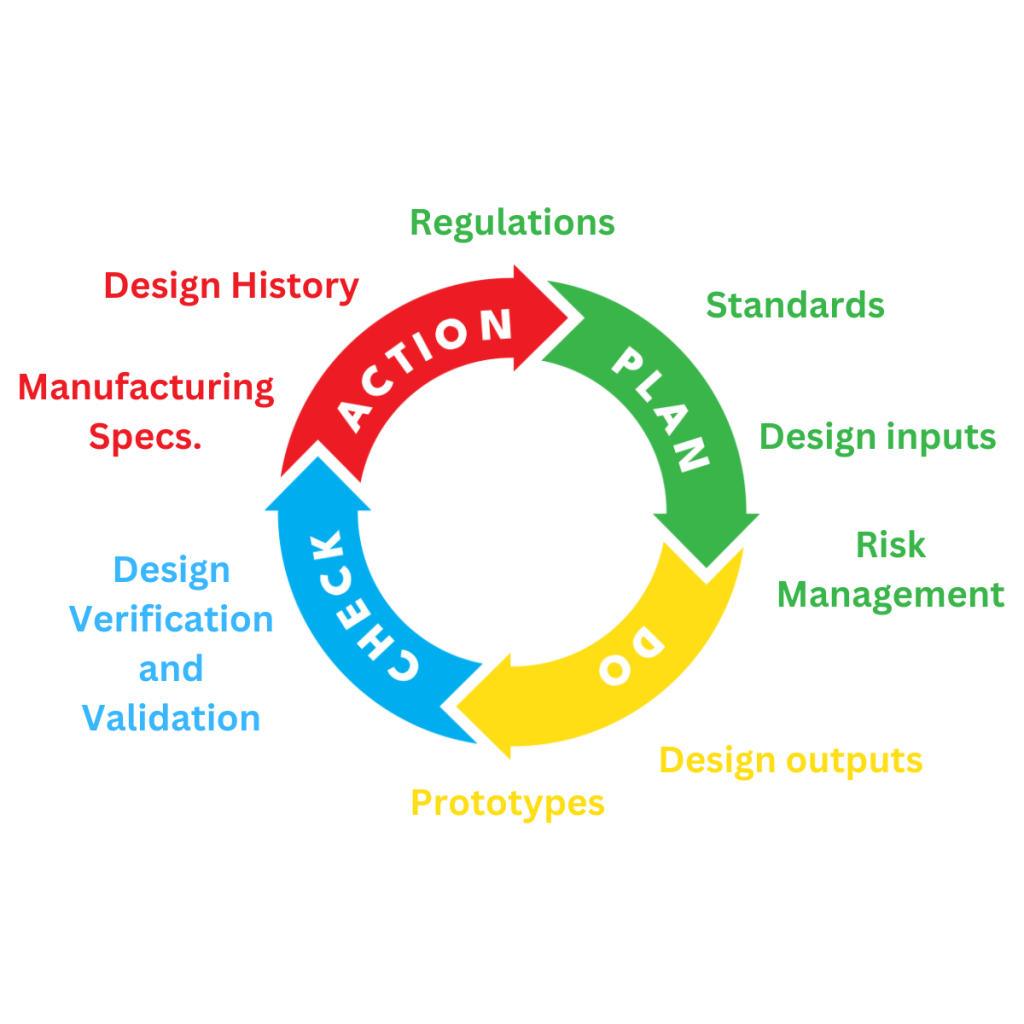
The Medical Device Design and Development Process
The medical device design and development process is a critical process that ensures
regulatory compliance and product safety. In this guide, we’ll share the lifecycle of medical device design
and development, highlighting relevant regulations, standards and documentation for product
manufacturers.

The Medical Device Design and Development Lifecycle
The lifecycle of the design and development of
medical devices and IVDs is meticulously
documented to ensure compliance and
traceability. In the following section, we’ll outline
the stages and documentation needed to meet
regulatory requirements.
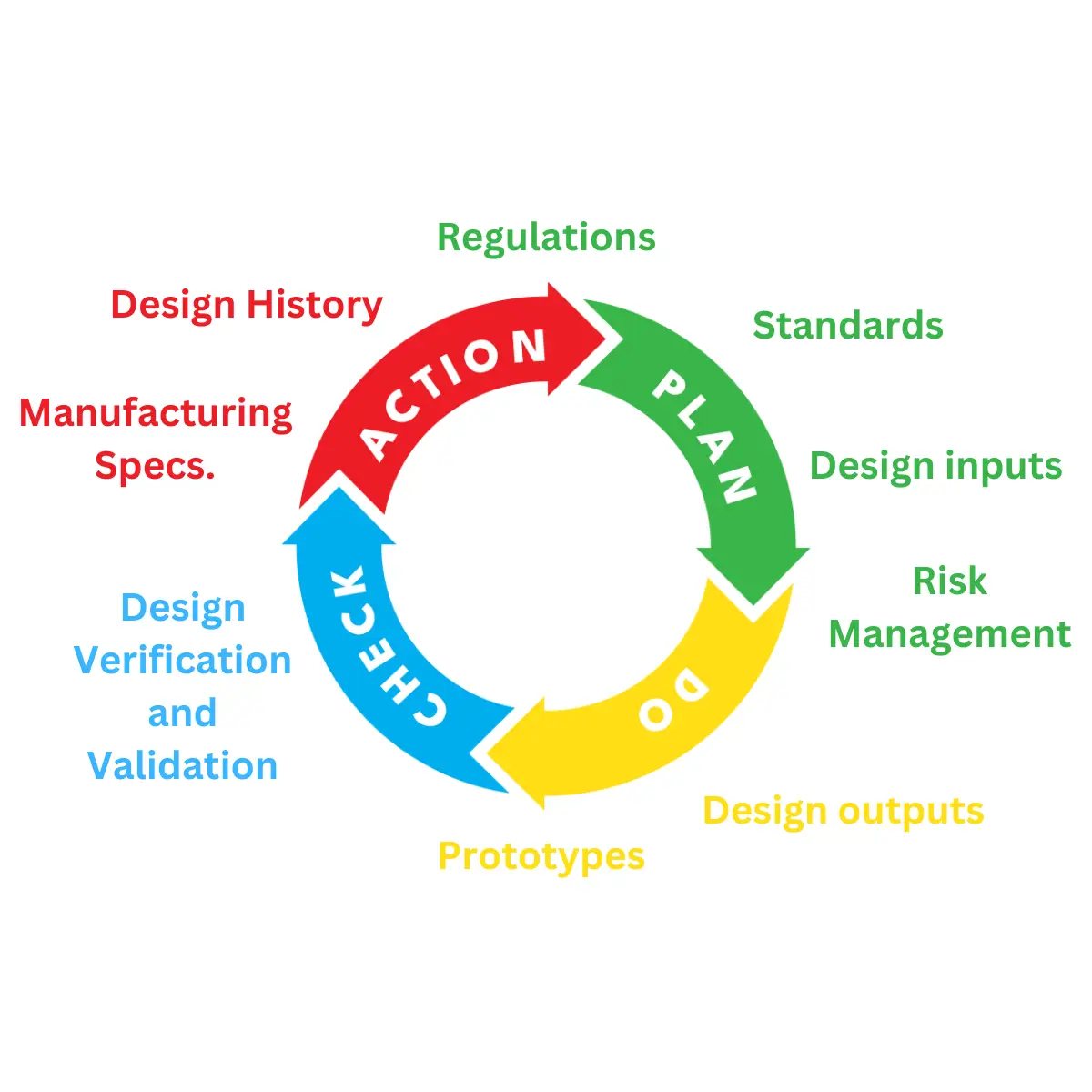
Medical Device and IVD Regulation
Regulations define the risk, safety, and performance requirements that must be met by manufacturers.
There are numerous different standards, which apply to different regions, groups and specific medical
devices. So, when you start to design and develop a medical device or IVD, it’s important to review the
regulations specific to its intended use. With an understanding of these, you can discuss them with the
design team, before they start the design and development process.
EU MDR and IVDR
In the European Union, the relevant regulations are:
- Medical devices – EU Medical Device Regulation (MDR) 2017/745
- IVDs – In Vitro Diagnostic Regulations (IVDR) (EU) 2017/746
These regulations outline the General Safety and Performance Requirements (GSPRs) in Annex I. These need to be assessed against the concept and intended use of the device being designed and developed:
UK Medical Device Regulations 2002
In the UK, compliance with the Medical Device Regulations 2002 is essential. Relevant regulations, which
outline the Essential Requirements (ERs) for design and development are:
- Medical devices – Medical Device Directives 93/42/EEC
- IVDs – 98/79/EC for IVDs.
The ERs can be found in Annex 1 of each of these directives, and should be assessed against the concept
and intended use of the medical device being developed.
International Medical Device Standards
Harmonised standards ensure that medical devices meet international safety and performance
benchmarks. Here are some important medical device-related standards that are accepted
internationally.
All medical devices:
- ISO 14971 – Medical devices. Application of risk management to medical devices
- ISO 20417 – Medical devices. Information to be supplied by the manufacturer
- ISO 15223 – Medical devices. Symbols to be used with information to be supplied by the manufacturer – General requirements
- ISO 13485 – Medical devices. Quality management systems. Requirements for regulatory purposes
- ISO 62366 – Medical devices – Application of usability engineering to medical devices
Medical devices containing software or software as a standalone medical device:
- IEC 62304 – Medical device software. Software life-cycle processes
Electrical devices:
- ISO 60601-1 series – General requirements for safety – Collateral standard. Safety requirements
for medical electrical systems - ISO 60601-2 series – Particular requirements for the basic safety and essential performance of magnetic resonance equipment for medical diagnosis
Biological Evaluation for patient contacting devices:
- ISO 10993 series – Evaluation and testing within a risk management process
Sterilised medical devices:
- ISO 11135 – Sterilization of health care products. Ethylene oxide – Requirements for
development, validation and routine control of a sterilization process for medical devices - ISO 11137 – Sterilization of health care products – Radiation – Requirements for development,
validation and routine control of a sterilization process for medical device - ISO 17665 – Sterilization of health care products – Moist heat – Requirements for the
development, validation and routine control of a sterilization process for medical devices - ISO 11607-1 – Packaging for terminally sterilized medical devices – Requirements for materials,
sterile barrier systems and packaging systems - ISO 11607-2 – Packaging for terminally sterilized medical devices – Validation requirements for
forming, sealing and assembly processes
For Specific Equipment:
- ISO 60601-2-24 – Medical electrical equipment – Particular requirements for the basic safety
and essential performance of infusion pumps and controllers
• ISO 3826-4 – Plastics collapsible containers for human blood and blood components –
Aphaeresis blood bag systems with integrated features
Design and Development Plan
Once you have a good understanding of the regulatory landscape, it is mandatory to develop a
comprehensive design and development plan. This should cover the stages of design, reviews,
verification, validation, and design transfer activities. It also includes responsibilities, authorities, and
traceability methods.

Quality Management Systems (QMS)
Before starting the medical device design and development process, it is important to commit to and maintain a Quality Management System (QMS) as laid
out in the following standards:
- USA – CFR Title 21 part 820 / EN ISO 13485
- EU – EN ISO 13485
The Design and Development process is set out within these QMS standards. And planning should cover
the following as a minimum.
https://www.ecfr.gov/current/title-21/chapter-I/subchapter-H/part-820/subpart-C/section-820.30
- Design and development stages
- Review(s) at each design and development stage
- Verification, validation, and design transfer activities appropriate to each design and development stage
- Responsibilities and authorities for design and development
- Methods ensuring traceability of design and development outputs to design and development inputs
- Necessary resources, including required experience / qualifications of personnel
Design and Development Stages
Designing medical devices involves several critical steps, each defined by EN ISO 13485 standards.
- Design Inputs include functional, performance, usability, and safety requirements, along with
relevant regulatory requirements and risk management outputs. - Design Outputs must meet input requirements, provide necessary information for purchasing, production and service provision, and specify product characteristics essential for safe use.
Design and Development Reviews
Systematic reviews ensure that design outputs meet requirements. These involve representatives from relevant functions and identify necessary actions required.
Design and Development Verification
Design Validation
Conducted on a representative product i.e., from initial production units, batches or equivalent, validation confirms that a product meets the requirements for its intended use.
It should also include methods, acceptance criteria, and rationalised statistical
techniques. This involves clinical evaluations or performance evaluations and is completed before product release.
Design and Development Transfer
Control of Design and Development Changes
Risk Management in Design and Development
Risk management is crucial throughout the medical device design and development process. In accordance with ISO 14971, risk management activities should be planned, executed, and documented throughout the product lifecycle.
Risk Management Plan
- Scope of planned risk management activities
- Assignment of responsibilities and authorities
- Requirements for reviewing risk management activity
- Criteria for risk acceptability
- Methods and criteria for evaluating overall residual risk
- Activities for verifying risk control measures
- Activities for reviewing production and post-production information
Risk Identification and Evaluation
Risks are identified at the start of the design and development process. They are then analysed and scored based on their likelihood and severity. Mitigation measures are then applied to reduce risks to acceptable levels.

Risk Control and Evaluation
Mitigated risks are re-evaluated to make sure they are within acceptable limits. A risk-benefit analysis then determines if the benefits of the device outweigh potential risks.
Mitigating Risk: An Example
Mitigation controls can include the use of standards. For example, a device which has patient contact and is sterilised using ethylene oxide.
Ethylene Oxide is known to cause irritation, organ damage, mutagenicity and carcinogenicity in human and animals, and reproductive effects in animals. So, the risk is ethylene oxide residuals on the device post-sterilisation and packaging, which could come into contact with the user.
Risk control measures would be to ensure that the ethylene oxide sterilisation process has been followed (ISO 11135) and testing carried out on the final finished post-sterilised device (ISO 10993-7).
Ethylene oxide sterilization residuals, and the test results evaluated as part of the Biological Evaluation Report by an expert toxicologist in line with ISO 10993-1 – Evaluation and testing within a risk management process.
The risk should then be scored again based on the control measure.
Proof of Concept
Proof of concept testing demonstrates that critical design features function as intended. This early-stage testing is crucial for identifying potential issues before clinical evaluations.
Design History
A comprehensive design history is maintained within the Quality Management System (QMS). This documentation includes detailed records of the design and development process.
By following these guidelines, medical device manufacturers can ensure that products meet regulatory requirements and are safe for use. This meticulous documentation and adherence to standards throughout design and development are critical for successful product development.
How can Patient Guard Help?
Patient Guard has helped hundreds of medical device and IVD manufacturers since 2017 with their medical device design and development. If you are designing and developing a medical device we can assist you with the regulatory requirements and technical documentation you need to provide as part of regulatory requirements. Contact us to see how we can assist with your medical device project.
FAQs
The design and development process for medical devices involves transforming an initial concept into a market-ready product. It includes defining user needs, creating a design plan, prototyping, testing, and ensuring regulatory compliance.
Why it matters: A structured process ensures the device is safe, effective, and meets regulatory requirements such as the EU MDR, FDA regulations, or ISO 13485.
The process typically includes:
- Concept and Feasibility: Defining user needs and technical feasibility.
- Design Planning: Developing a structured plan, including regulatory considerations.
- Design Inputs: Documenting specifications based on user needs and safety requirements.
- Design Outputs: Creating detailed designs, including prototypes and schematics.
- Design Verification and Validation: Ensuring the design meets specifications and performs as intended.
- Transfer to Manufacturing: Scaling production with quality controls in place.
Pro tip: Aligning the process with regulatory standards like ISO 13485 ensures smoother market entry.
Risk management, as outlined in ISO 14971, is integral to medical device design. It involves:
- Identifying Risks: Assessing potential hazards associated with the device.
- Mitigating Risks: Designing controls to minimize risks.
- Residual Risk Evaluation: Ensuring remaining risks are acceptable.
- Ongoing Monitoring: Updating risk assessments based on post-market data.
Key takeaway: Incorporating risk management early reduces safety issues and ensures compliance.
- Design Verification: Confirms the design meets input specifications (e.g., performance, safety).
- Design Validation: Ensures the device meets user needs and intended purposes under real-world conditions.
Why it matters: Verification and validation are regulatory requirements under frameworks like FDA QSR and EU MDR, and they ensure the device functions safely and effectively.
Regulations like the EU MDR, IVDR, and FDA guidelines shape every stage of design and development. Key requirements include:
- Defining intended use and classification early in the process.
- Ensuring a Quality Management System (QMS) aligned with ISO 13485.
- Documenting design controls, risk management, and verification/validation results.
- Preparing for regulatory submissions with complete technical documentation.
Key insight: Integrating compliance from the start avoids costly redesigns or approval delays.
Yes! Patient Guard offers expert support throughout the design and development process, including:
- Developing regulatory-compliant design and development plans.
- Assisting with risk management and design verification/validation.
- Preparing technical documentation for CE marking, FDA, or UKCA submissions.
- Guiding compliance with standards like ISO 13485 and ISO 14971.
Why choose Patient Guard: With extensive experience in regulatory frameworks and over 500 successful projects, we ensure your device is designed for safety, performance, and compliance.

