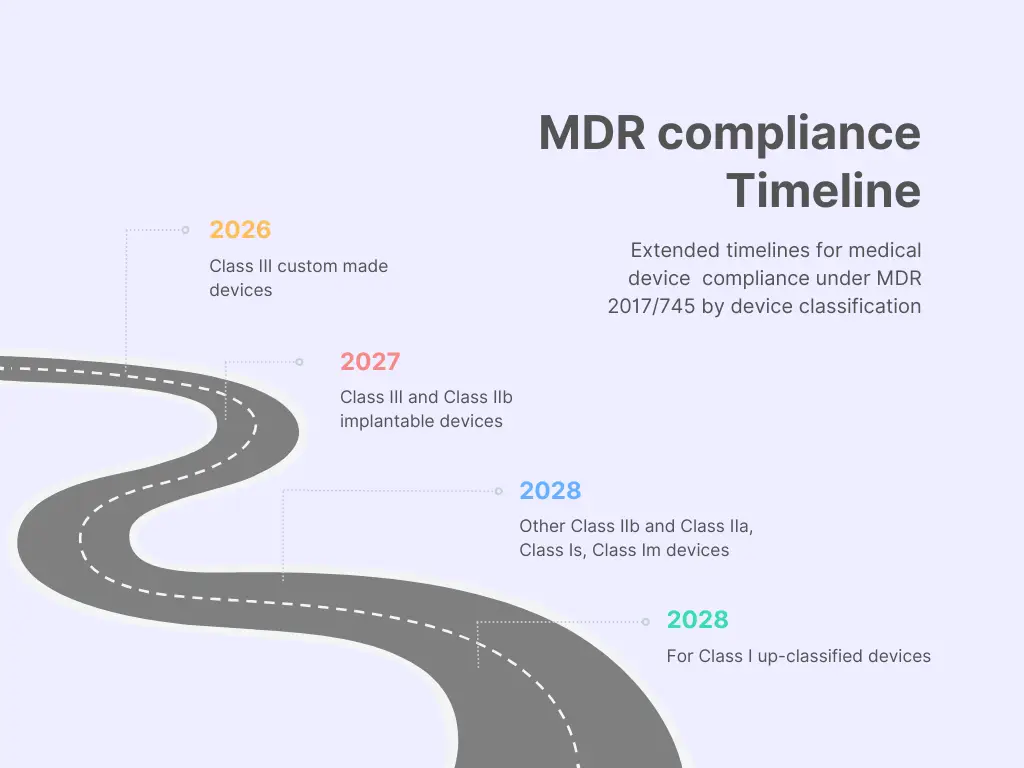
MDR Article 120
In the fast-paced world of medical device manufacturing, regulatory standards are the bedrock of patient safety and product quality. Recently, the European Union has taken a significant step by extending Article 120 of the EU Regulation 2017/745 and EU Regulation 2017/746. This move has been made to address the mounting backlog of CE mark certifications and re-certifications within notified bodies, the entities responsible for ensuring that medical devices meet stringent quality and safety requirements before they reach the market. In this blog post, we’ll delve into the implications of this extension for medical device manufacturers and emphasize the critical importance of continued compliance.